
Eur J Case Rep Intern Med. 2025 Dec 4;12(12):005837. doi: 10.12890/2025_005837. PMID: 41377782; PMCID: PMC12688513.
POINTS À RETENIR
-
Le syndrome d'Ehlers-Danlos vasculaire (vEDS) est une maladie héréditaire rare du tissu conjonctif caractérisée par une fragilité artérielle extrême, nécessitant une suspicion clinique accentuée pour éviter les erreurs de diagnostic et les interventions inappropriées.
-
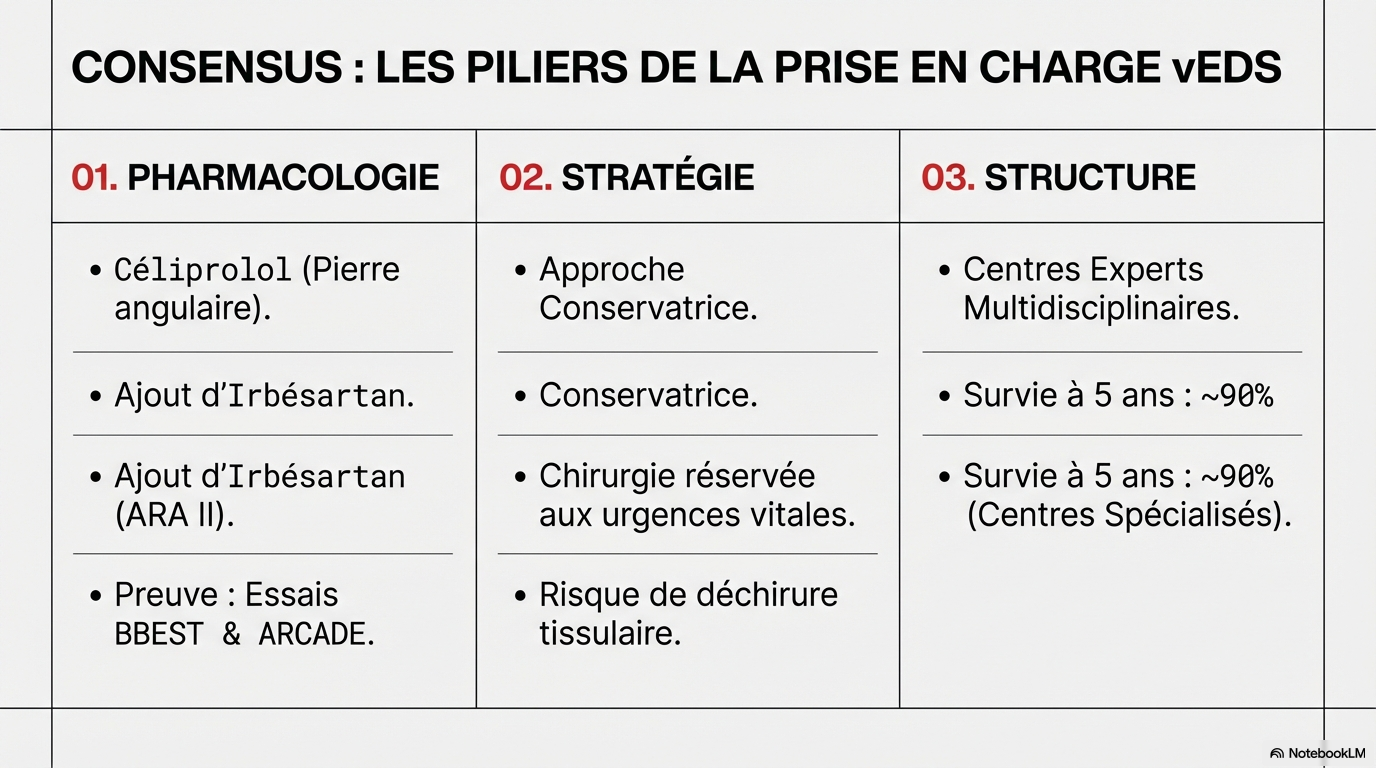
Le strict respect des traitements pharmacologiques fondés sur des données probantes et un suivi structuré à long terme sont essentiels, car l'arrêt du traitement peut entraîner des événements vasculaires rapides et potentiellement mortels.
-
La prise en charge devrait être centralisée dans des centres experts adoptant une approche multidisciplinaire, où des stratégies individualisées et majoritairement conservatrices peuvent optimiser les résultats chez cette population à haut risque.
Critères diagnostiques par type principal
Utilisez la classification de 2017 (critères de New York)
-

Cette étude clinique explore les complexités médicales et les défis de prise en charge liés au syndrome d’Ehlers-Danlos vasculaire (vEDS), une maladie génétique rare entraînant une fragilité artérielle extrême. À travers l'analyse de trois cas concrets, les auteurs soulignent l'importance vitale d'une adhérence stricte au traitement pharmacologique, notamment au céliprolol, pour prévenir des complications mortelles. Le texte met en lumière la nécessité d'un suivi multidisciplinaire dans des centres experts afin d'éviter les erreurs de diagnostic et de privilégier des stratégies conservatrices face aux risques élevés des interventions chirurgicales. En fin de compte, cet article sert de plaidoyer pour une surveillance continue et coordonnée tout en reconnaissant le besoin urgent de nouvelles options thérapeutiques pour améliorer le pronostic vital de ces patients.

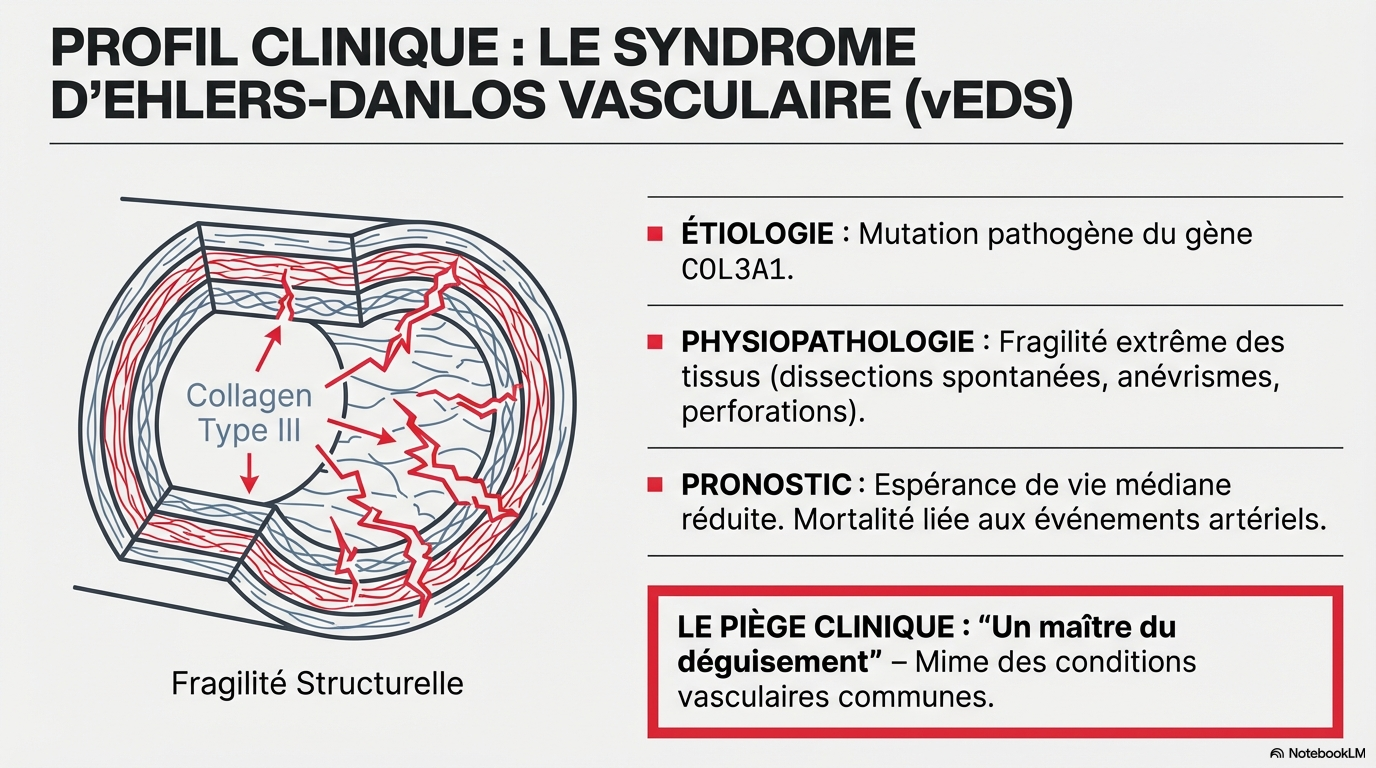
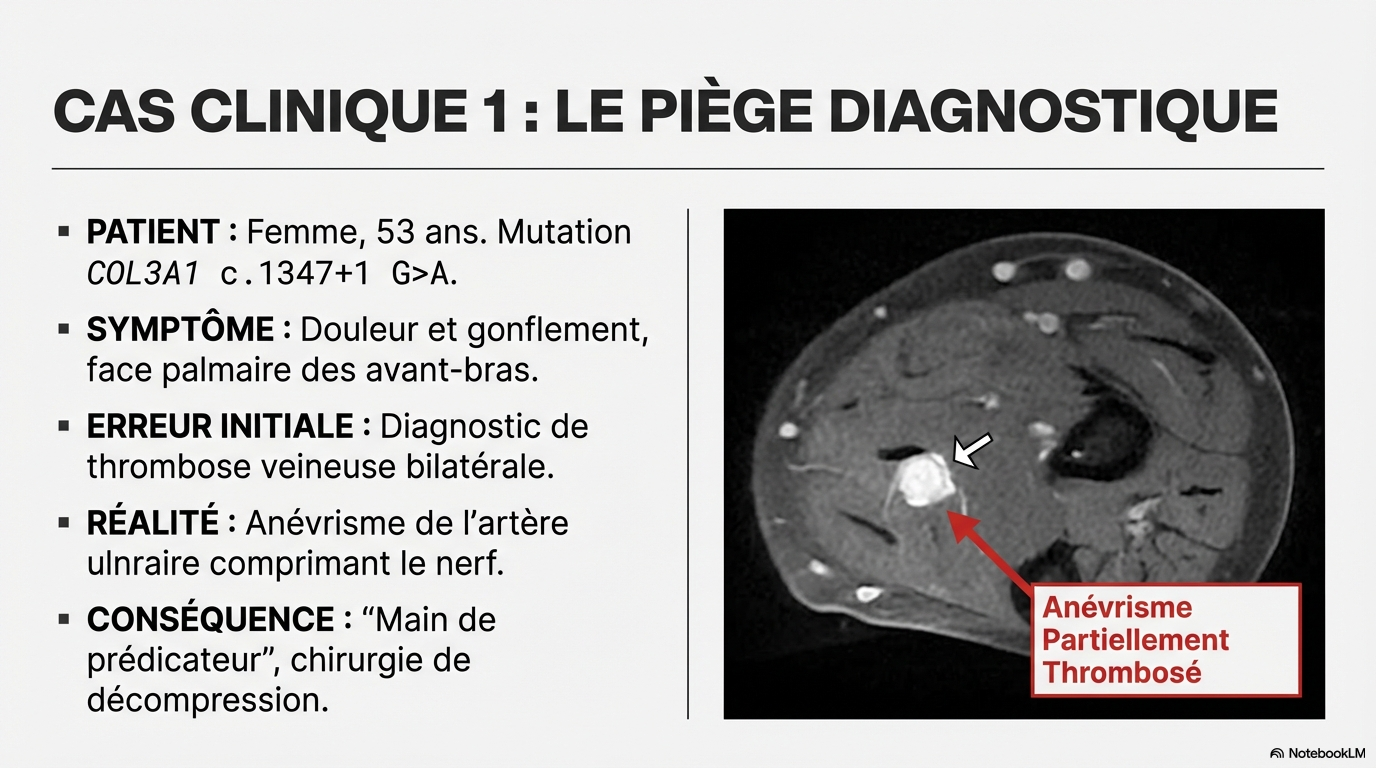
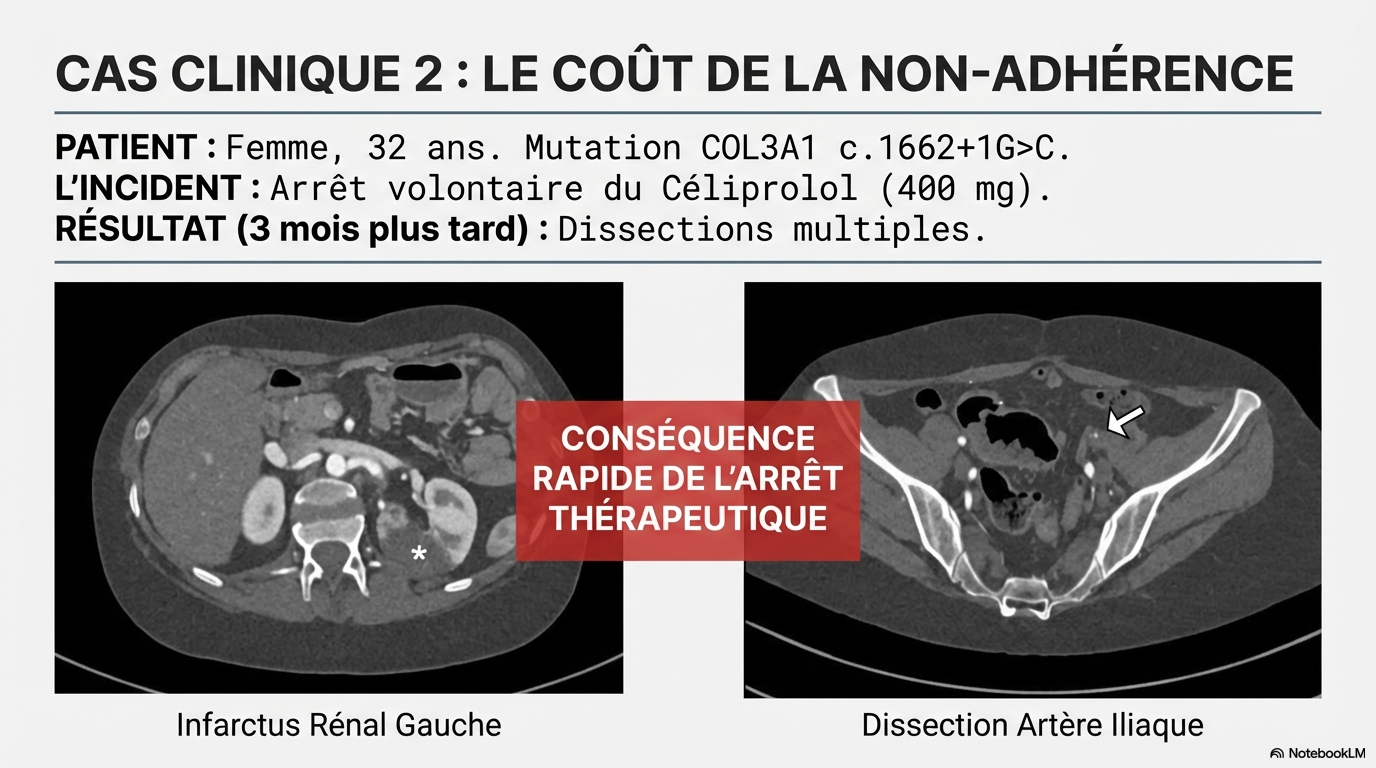
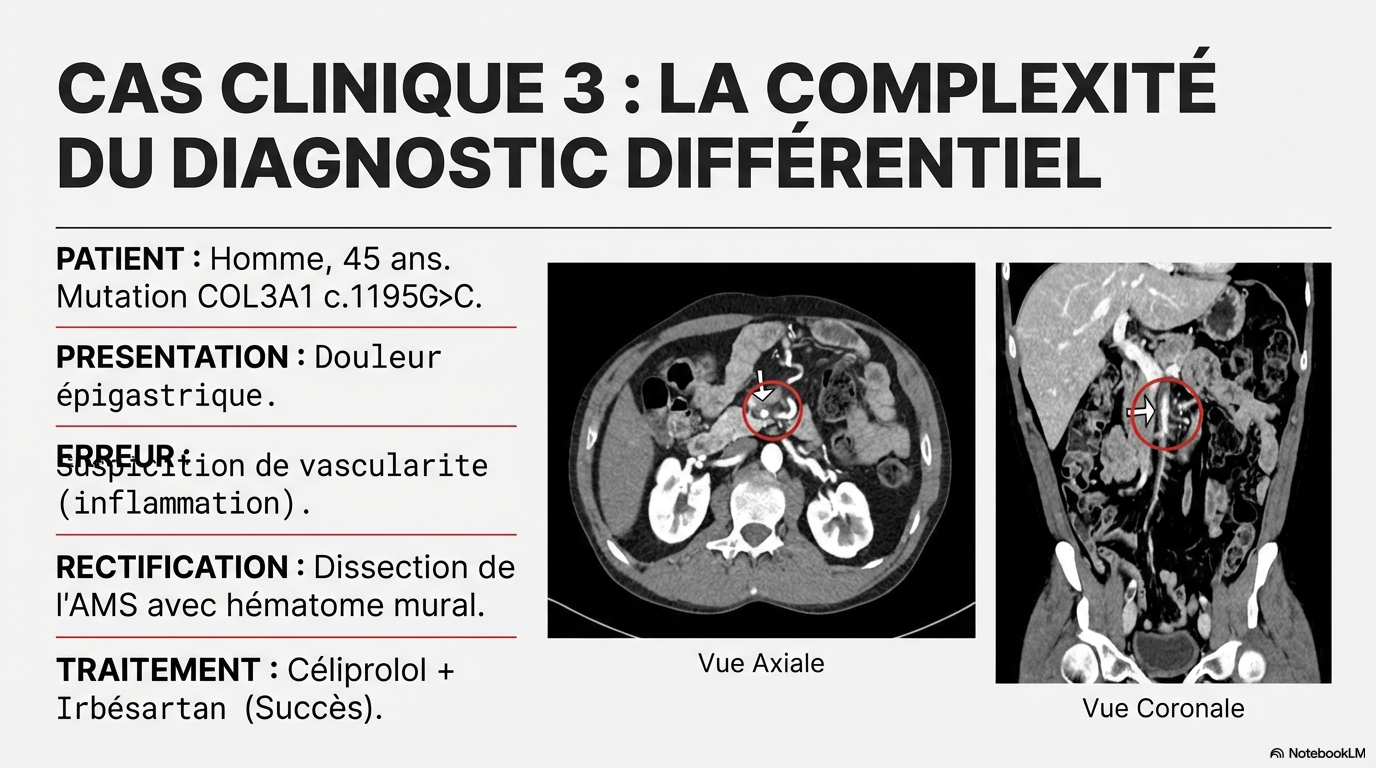

Ne pas passer à côté, y penser !
Arbre généalogique indispensable

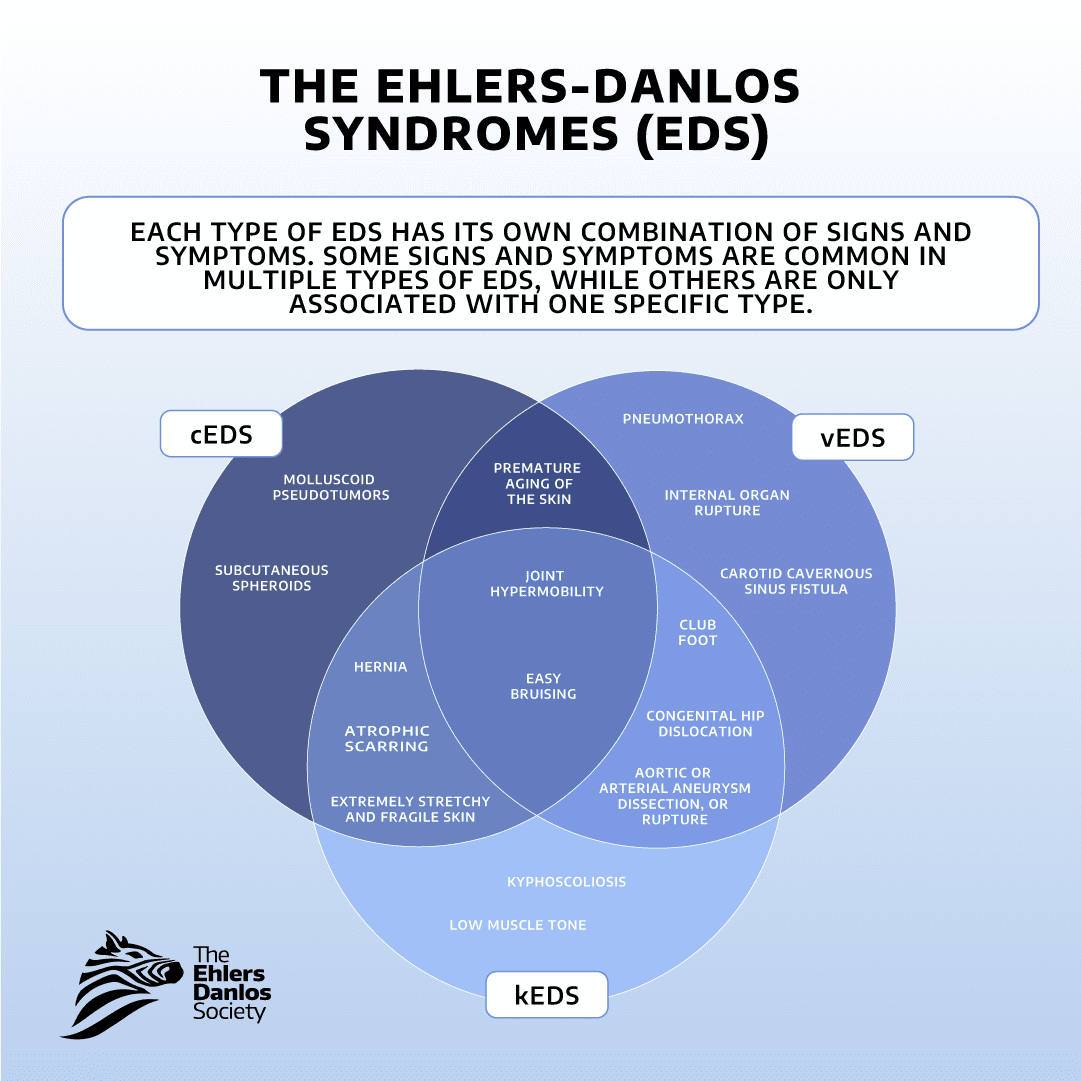
La classification actuelle comprend 13 types de syndrome d'Ehlers-Danlos. Chaque type a ses propres causes génétiques, ce qui crée un ensemble unique de caractéristiques pour chaque type de SED.
Fréquence
Chaque type de SED a une prévalence différente au sein de la population. Le SED hypermobile (SEDh) est de loin le plus fréquent. Il représente environ 90 % des cas de SED et toucherait au moins une personne sur 1 3,100 à 5,000 . Le SEDh est actuellement classé comme une maladie rare, mais sa prévalence réelle est inconnue et pourrait être sous-estimée. Le SED classique (SEDc) et le SED vasculaire (SEDv) sont beaucoup plus rares que le SEDh. Le SEDc touche environ une personne sur 20,000 40,000 à 1 100,000. Le SEDv touche environ une personne sur 200,000 1 à 1 XNUMX. Tous les autres types de SED sont classés comme ultra-rares, touchant moins d'une personne sur XNUMX million. Plusieurs types de SED n'ont été signalés que dans quelques familles touchées.
Les syndromes d'Ehlers-Danlos peuvent être transmis des parents à l'enfant. Chaque type de syndrome d'Ehlers-Danlos est transmis selon un mode dominant ou récessif.
Un mode de transmission dominant signifie qu'une seule copie d'une variante génétique (transmise par un parent) est nécessaire pour hériter de la maladie. Si une personne est atteinte d'une maladie à mode de transmission dominant, chacun de ses enfants aura 50 % de chances d'en hériter. Un mode de transmission récessif signifie qu'une personne doit hériter de deux copies de la variante génétique (une de chaque parent) pour être atteinte de la maladie.
Bien que les causes génétiques du SEDh ne soient pas encore connues, les antécédents familiaux suggèrent que cette maladie a un mode de transmission dominant.
Chaque type de SED possède ses propres critères diagnostiques cliniques. Ces critères sont un ensemble de symptômes et de caractéristiques observés dans chaque affection. Si une personne répond aux critères de diagnostic pour un type de SED, un test génétique doit être effectué pour confirmer le diagnostic.
La ou les causes génétiques du SEDh n'ont pas encore été identifiées ; il n'existe donc actuellement aucun test génétique permettant de le diagnostiquer. Le diagnostic de SEDh est posé chez les personnes répondant aux critères cliniques.
Si une personne présente une hypermobilité articulaire symptomatique, mais ne répond pas aux critères diagnostiques d'un SED ou d'une autre affection pouvant entraîner des symptômes similaires, il convient d'envisager un trouble du spectre de l'hypermobilité (TSH).
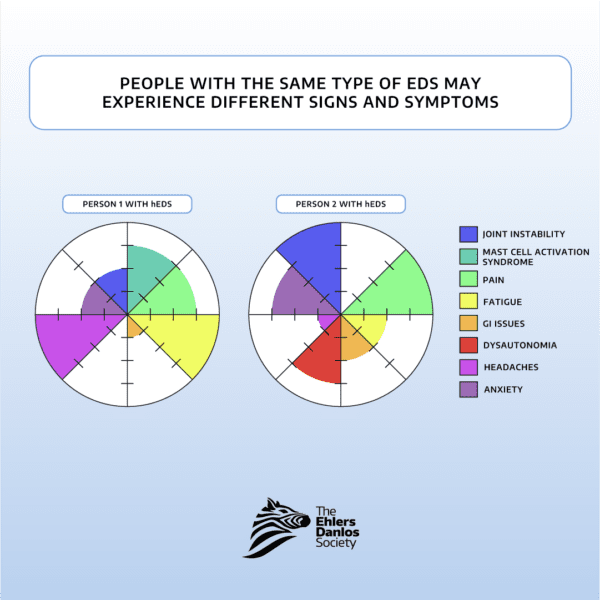
Il n'existe pas de traitement spécifique pour chaque type de SED. La prise en charge du SED repose donc sur celle des symptômes de chaque personne. Les syndromes d'Ehlers-Danlos peuvent provoquer divers symptômes dans différentes parties du corps. Par conséquent, les personnes atteintes d'un même type de SED nécessitent souvent la prise en charge de plusieurs professionnels de santé. Même au sein d'un même type de SED, deux personnes peuvent présenter des symptômes très différents et réagir différemment à des stratégies de prise en charge différentes. Chaque personne doit collaborer avec son équipe soignante pour élaborer un plan de soins adapté à ses besoins spécifiques.
https://www.ehlers-danlos.com/fr/l%27%C3%A9v%C3%A8nementiel/symposium-scientifique-international-2025/

Syndrome d'Ehlers-Danlos vasculaire
"Qu’est-ce qu’un syndrome d’Ehlers-Danlos vasculaire ?
Il existe différentes formes de syndromes d'Ehlers Danlos, dont le syndrome d’Ehlers Danlos vasculaire (anciennement type IV) qui est une maladie génétique rare (1 patient / 150 000) due à des mutations hétérozygotes du gène COL3A1 touchant le collagène de type III. Sa transmission est autosomique dominante, non liée au sexe puisque le gène COL3A1 se situe sur le chromosome 2.
Comme la plupart des gènes, le gène COL3A1 est présent en 2 exemplaires dans notre génome. Il suffit qu’une des 2 copies soit défectueuse pour que la production de collagène de type III soit altérée et pour que la maladie soit possible.
Le collagène de type III est particulièrement présent dans les vaisseaux (artères et veines), les intestins, la peau, l'utérus, mais également dans les poumons, le foie, la rate et les capsules articulaires."
https://www.maladies-vasculaires-rares.fr/syndrome-dehlers-danlos-vasculaire
