« Notae vero inflammationis sunt quatuor : rubor et tumor cum calore et dolore. » (Les quatre signes de l'inflammation sont : la rougeur et le gonflement, avec la chaleur et la douleur.) — Celse (Aulus Cornelius Celsus) dans De Medicina.
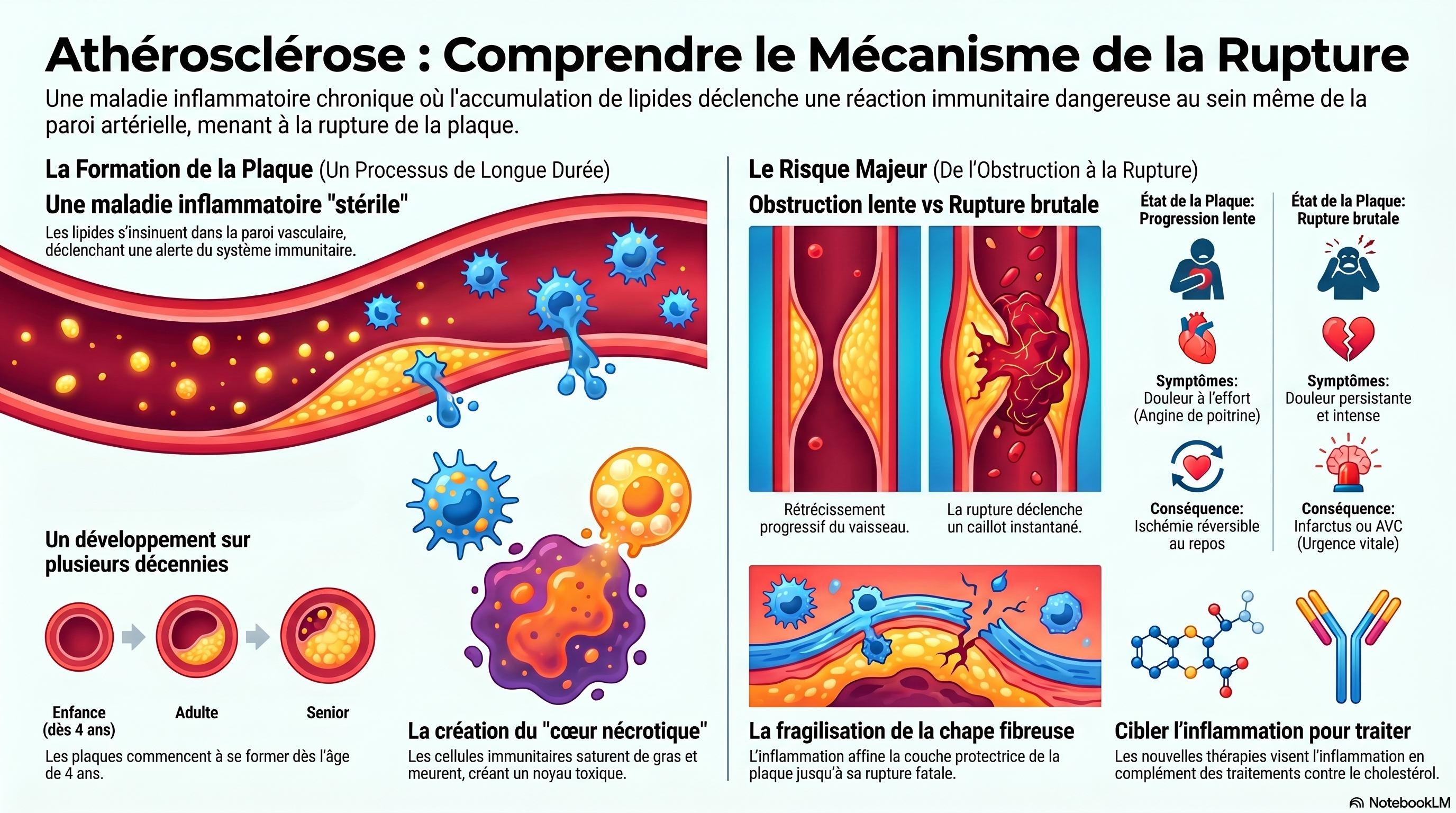
« L'athérosclérose n'est plus considérée comme un simple problème de "plomberie" avec une accumulation passive de cholestérol, mais comme une véritable maladie inflammatoire chronique de la paroi artérielle. » — Concept central issu des travaux du Dr Peter Libby et des pionniers de la biologie vasculaire. C'est cette inflammation qui fragilise la plaque d'athérome et conduit à la thrombose.

https://pmc.ncbi.nlm.nih.gov/articles/PMC11555284/
Faits
-
La physiopathologie de l'athérosclérose est étroitement liée à l'inflammation et à sa transition vers une inflammation chronique.
-
Les monocytes/macrophages sont au cœur de l'inflammation des plaques dans l'athérosclérose, avec des mécanismes moléculaires vastes et complexes qui ne sont pas entièrement décryptés.
-
Les découvertes moléculaires actuelles sur l'inflammation athéroscléreuse ont peu de chances d'être transposées en clinique.
-
Questions ouvertes
-
Pourquoi la transposition thérapeutique des cibles moléculaires identifiées dans l'inflammation athéroscléreuse est-elle limitée ?
-
Comment exploiter certains des mécanismes endogènes activés au sein des macrophages qui semblent être anti-inflammatoires et athéroprotecteurs ?
-
Pourquoi la découverte des mécanismes moléculaires de l'inflammation dans l'athérosclérose devrait-elle être privilégiée par rapport aux interventions thérapeutiques hâtives (souvent confondues avec des stratégies innovantes) ?
EXTRAITS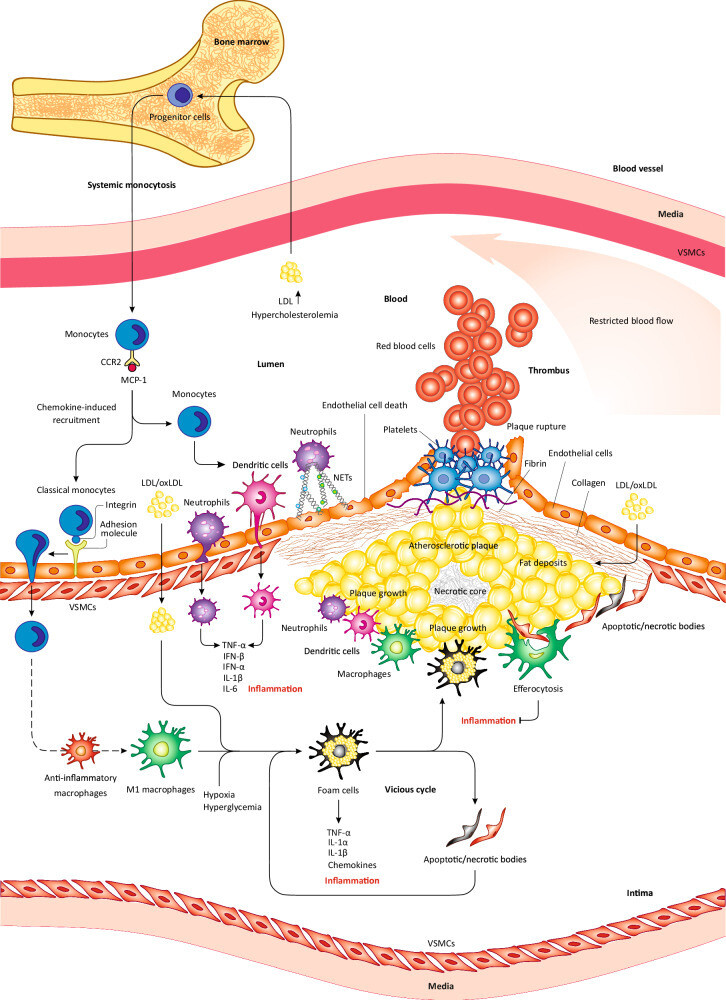
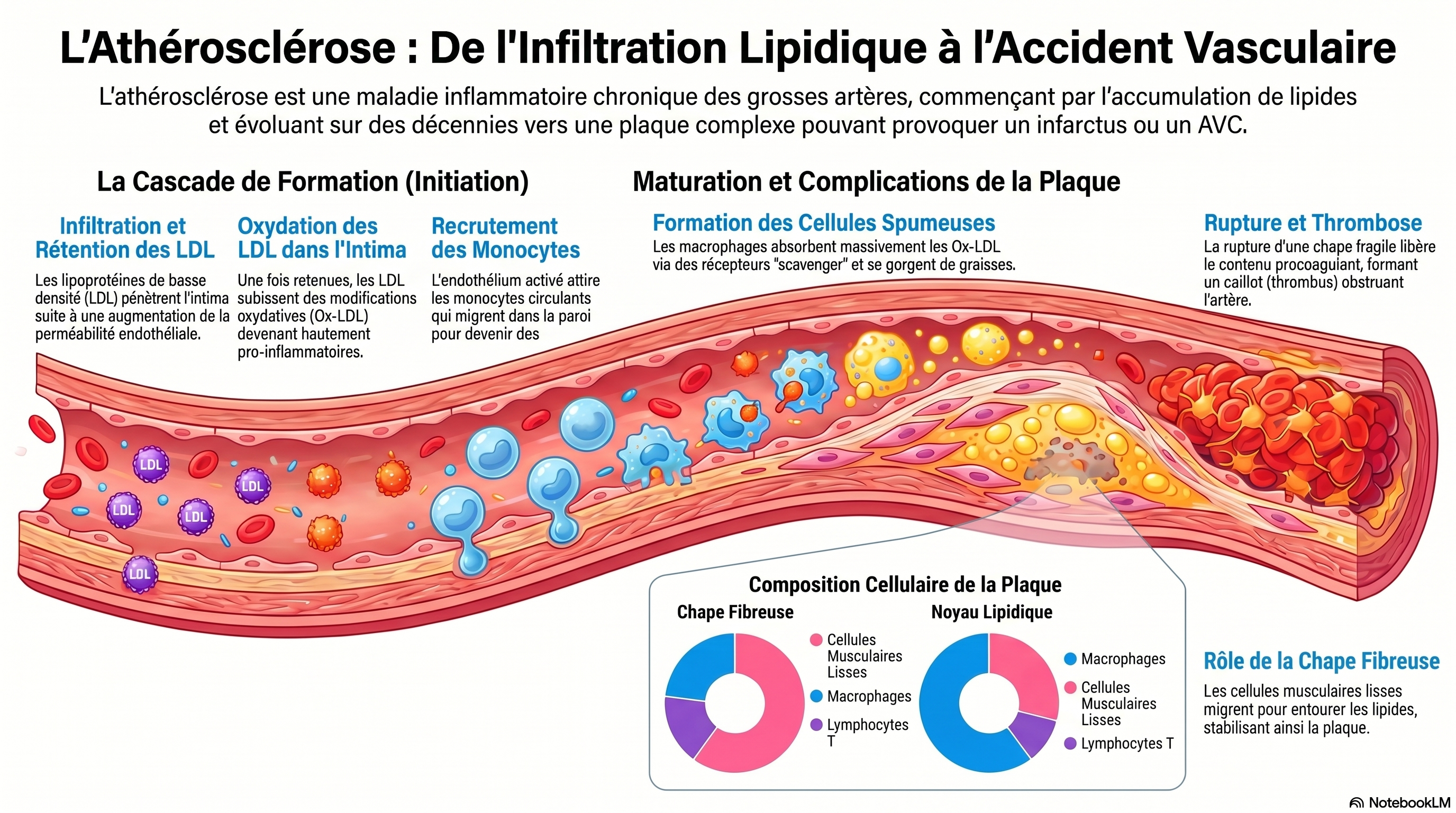
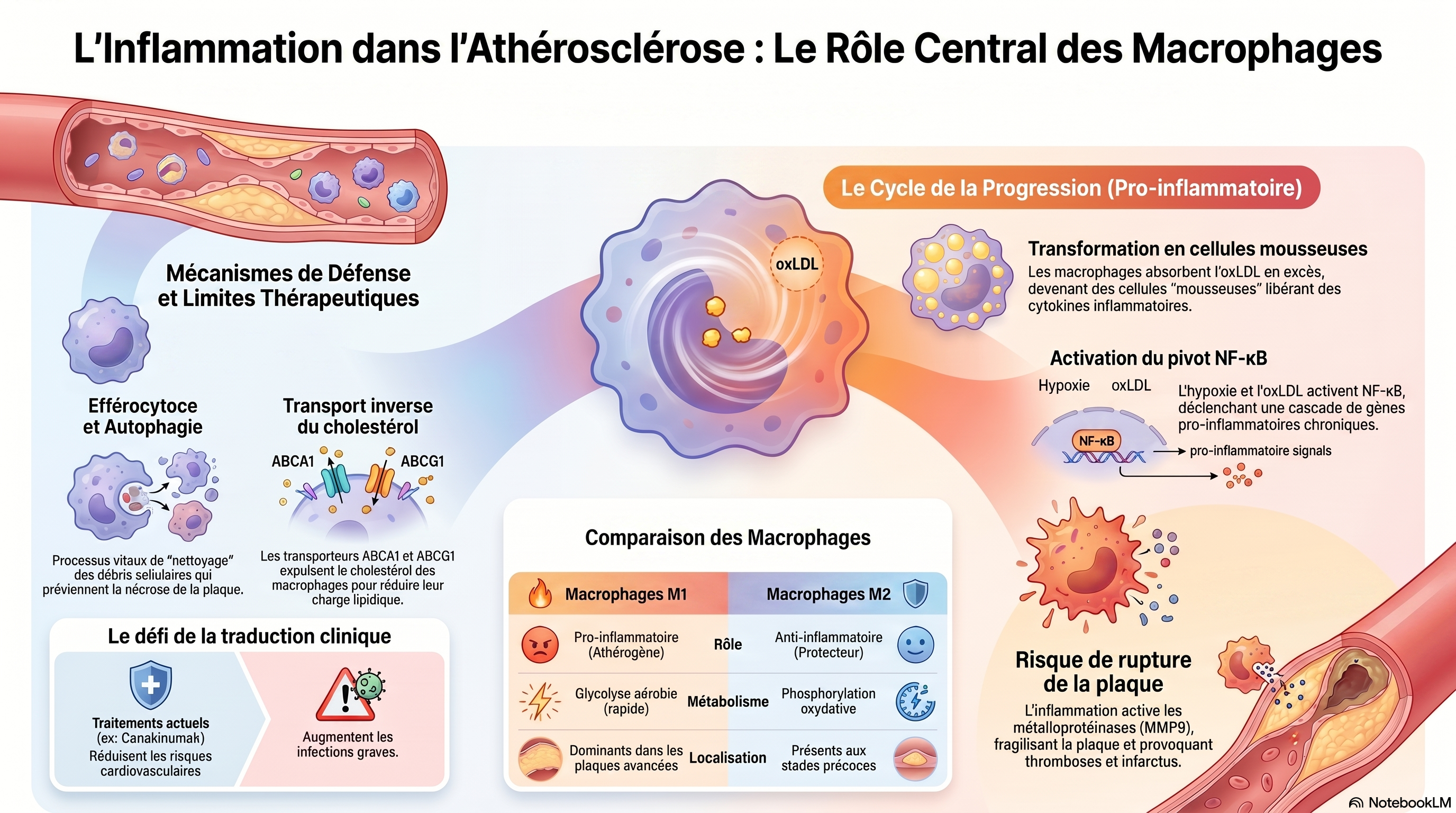
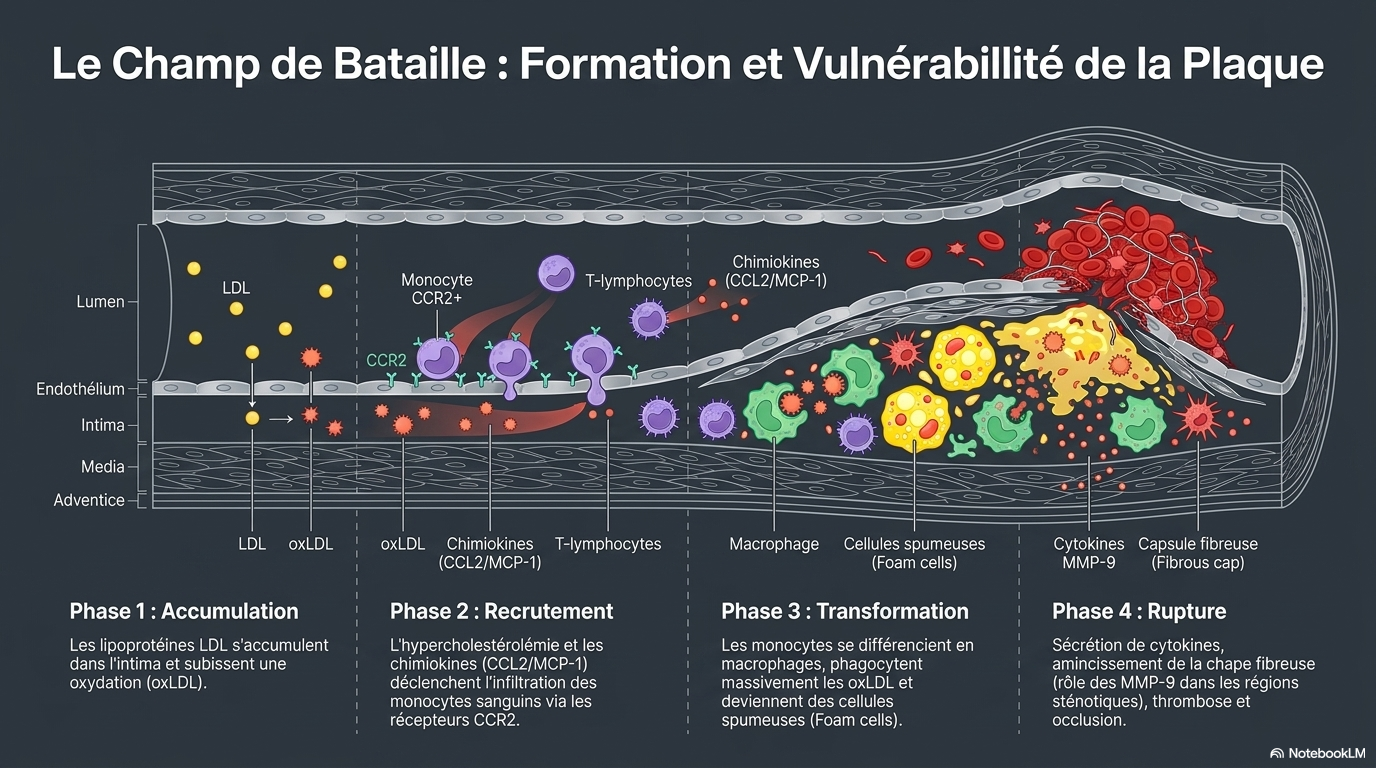
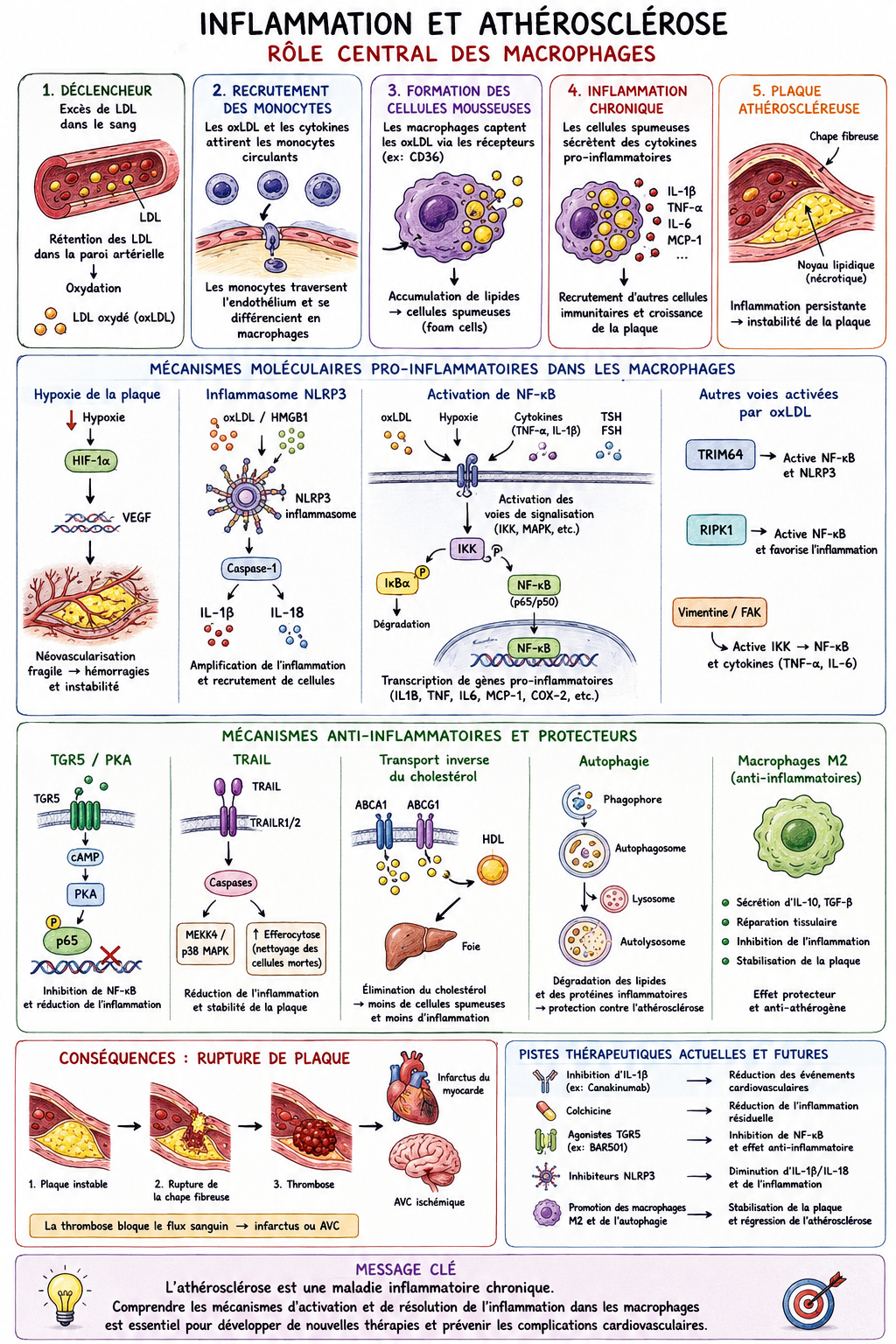
La rétention ou l'oxydation excessive du LDL dans la couche sous-endothéliale artérielle induit la génération de monocytes à partir de cellules progénitrices de la moelle osseuse et leur libération subséquente dans la circulation. Sous l'influence de chimiokines comme le MCP-1, les monocytes circulants migrent vers les lésions/plaques athéroscléreuses inflammatoires, où ils se différencient en cellules dendritiques ou en macrophages après infiltration de l'endothélium. Les macrophages inflammatoires libèrent des chimiokines/cytokines qui favorisent l'inflammation de la plaque. Initialement, les macrophages contribuent à réduire la charge en LDL ; cependant, face à une accumulation excessive de LDL oxydées (oxLDL), ils se transforment en cellules spumeuses qui libèrent davantage de cytokines/chimiokines, aggravant ainsi l'inflammation, ou se déposent dans la plaque, entraînant sa croissance. À mesure que la plaque grossit, elle devient instable et peut se rompre. En raison du contexte inflammatoire de la plaque, des facteurs procoagulants sont activés et la production de fibrine augmente. L'ensemble de ces modifications provoque la coagulation sanguine au site de rupture et la formation d'un thrombus. De plus, la nécrose des cellules spumeuses et d'autres cellules inflammatoires contribuent à l'accumulation du noyau nécrotique. Si l'efférocytose prévient l'inflammation et la croissance de la plaque, son altération au niveau des corps apoptotiques/nécrotiques peut entraîner un dépôt accru de macrophages/cellules spumeuses dans la plaque. En raison de l'inflammation présente dans la plaque, les monocytes se différencient en cellules dendritiques (DC), qui infiltrent l'endothélium et libèrent des cytokines pro-inflammatoires favorisant l'inflammation et l'athérosclérose. De même, sous l'effet de la stimulation par des médiateurs inflammatoires, les neutrophiles se différencient et infiltrent la plaque à travers l'endothélium, où ils libèrent des cytokines pro-inflammatoires qui favorisent également l'inflammation et l'athérosclérose. Les NET libérés par les neutrophiles induisent également des lésions endothéliales et la mort cellulaire, ce qui peut conduire à la rupture de la plaque.
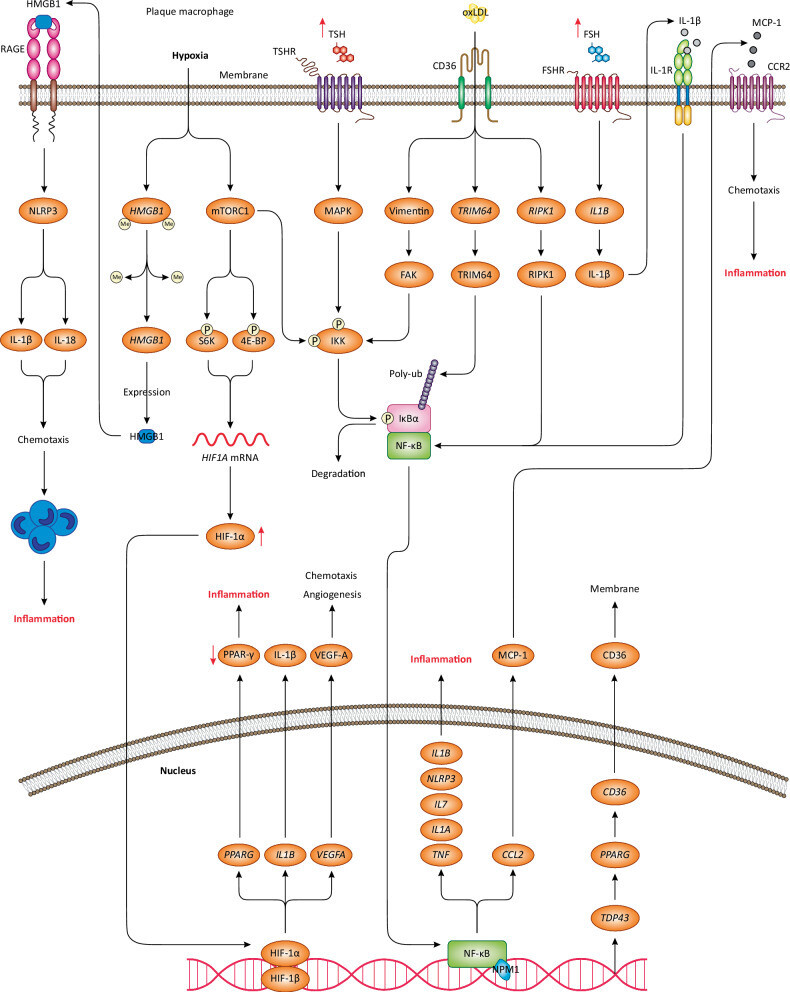
-
Lors d'une hypoxie de la plaque d'athérome, deux voies de signalisation principales sont activées dans les macrophages : mTORC1 et HMGB1. L'augmentation de la transcription et de l'activation de HMGB1 stimule les récepteurs RAGE à la surface des macrophages, entraînant l'activation de l'inflammasome NLRP3. Ce dernier libère les chimiokines IL-1β et IL-18, induisant la chimiotaxie et le recrutement des monocytes/macrophages au niveau de la plaque, et favorisant ainsi l'inflammation. L'activation de mTORC1 augmente également la transcription et l'expression de HIF1A /HIF-1α, qui migre vers le noyau et stimule la transcription de plusieurs gènes, jouant un rôle clé dans l'angiogenèse et la chimiotaxie, et induisant ainsi l'inflammation de la plaque. De plus, mTORC1 phosphoryle et active le complexe IKK, qui phosphoryle IκBα, entraînant sa dégradation et sa dissociation de NF-κB, un facteur de transcription essentiel à la régulation de l'inflammation par les macrophages. NF-κB migre vers le noyau et stimule la transcription de gènes pro-inflammatoires clés, de cytokines et de chimiokines, déclenchant ainsi l'inflammation. CCL2 /MCP-1 est une chimiokine essentielle qui se lie au récepteur CCR2 des macrophages, induisant la chimiotaxie et l'inflammation. Un taux plasmatique de TSH anormalement élevé active la voie de signalisation MAPK après sa liaison au récepteur TSHR des macrophages. Cette voie MAPK conduit à la phosphorylation du complexe IKK, qui active NF-κB. De même, un taux plasmatique élevé de FSH active le récepteur FSHR des macrophages, entraînant une augmentation de la transcription, de l'activation et de la libération d' IL -1β, qui se lie ensuite au récepteur IL-1R des macrophages, amplifiant ainsi l'activation de NF-κB. Par des mécanismes encore inconnus, l'expression de TDP43 est augmentée dans les macrophages, ce qui stimule la transcription et l'activation de CD36 /récepteur scavenger CD36. CD36 se transloque à la membrane et permet l'internalisation des oxLDL/LDL. Ceci entraîne l'activation de la vimentine, qui induit la phosphorylation du complexe IKK par l'activation de la kinase FAK, conduisant à l'activation de NF-κB. De plus, l'internalisation des oxLDL est associée à une augmentation de la transcription et de l'activation de TRIM64 /TRIM64 et RIPK1 /RIPK1, qui interviennent dans l'activation de NF-κB. Compte tenu du rôle central de NF-κB dans la réponse pro-inflammatoire des macrophages, son activation excessive conduit à une inflammation chronique au sein de la plaque d'athérome.
Conclusion et perspectives d'avenir
Les macrophages jouent un rôle prépondérant dans l'athérosclérose et l'inflammation chronique des plaques.
Dans cette revue, nous nous sommes intéressés aux mécanismes moléculaires et aux voies de signalisation de l'inflammation au sein des macrophages des lésions/plaques athérosclérotiques. Nous avons décrit comment l'activation de ces mécanismes induit une inflammation qui évolue ensuite vers une inflammation chronique des plaques, aggravant ainsi l'athérosclérose.
L'athérosclérose peut donc être perçue comme un cercle vicieux qui amplifie l'inflammation, car la plupart des événements survenant lors de sa physiopathologie (par exemple, le recrutement/l'activation cumulatif des monocytes/macrophages et d'autres cellules immunitaires, la présence d'hypoxie et d'oxLDL, l'angiogenèse accrue, etc.) tendent à exacerber certains aspects ou modes d'inflammation.
Bien qu'une discussion exhaustive des mécanismes de l'inflammation dans l'athérosclérose nécessiterait des revues de littérature supplémentaires, nous avons concentré notre analyse sur les études majeures de ces dernières années.
Nous nous sommes intéressés à des éléments clés tels que l'hypoxie, l'absorption des oxLDL et des LDL, les modifications hormonales, les facteurs de transcription, les cytokines et les chimiokines, ainsi que le rôle crucial de l'activation de NF-κB dans l'inflammation. Lorsque cela était possible, nous avons mis en lumière les cibles moléculaires potentielles, les avantages et les inconvénients de leur ciblage, et leurs applications thérapeutiques potentielles.
Nous concluons qu'en raison de l'immense complexité et des mécanismes encore largement inexplorés de l'inflammation dans l'athérosclérose, les cibles moléculaires efficaces pour le développement de stratégies thérapeutiques sont limitées.
Par conséquent, l'inflammation moléculaire dans l'athérosclérose est un domaine émergent, riche de nouvelles découvertes chaque année.
Nous espérons pouvoir un jour maîtriser ces mécanismes et réduire l'inflammation dans l'athérosclérose, ce qui pourrait atténuer significativement la maladie. De plus, comme décrit précédemment, certains mécanismes sont activés dans les macrophages, ce qui pourrait s'avérer prometteur pour la prévention de l'inflammation athérosclérotique. Cependant, des études complémentaires sont nécessaires pour confirmer la validité de ces mécanismes et leurs applications thérapeutiques.
De manière générale, nous pensons qu'il convient de donner la priorité à l'approfondissement de nos connaissances sur les mécanismes moléculaires fondamentaux et les cascades de signalisation de l'inflammation dans l'athérosclérose afin de développer des interventions thérapeutiques efficaces.
Cet article de synthèse scientifique examine le rôle fondamental de l'inflammation dans l'athérosclérose, soulignant que cette pathologie n'est pas qu'un simple stockage de graisses, mais un processus immunitaire complexe. Le texte s'articule autour du comportement des macrophages, qui, après avoir absorbé un excès de cholestérol LDL, se transforment en cellules foam (cellules écumeuses) et déclenchent des cascades de signalisation pro-inflammatoires via des facteurs comme le NF-κB et l'inflammasome NLRP3. Les auteurs explorent des thèmes cruciaux tels que l'impact de l'hypoxie sur la stabilité des plaques et les mécanismes de l'efférocytose, un processus de nettoyage cellulaire souvent défaillant dans les stades avancés de la maladie. Enfin, l'objectif est d'identifier de nouvelles cibles thérapeutiques translationnelles, comme la polarisation des macrophages vers le phénotype anti-inflammatoire M2 ou l'activation de l'autophagie, afin de freiner la progression des maladies cardiovasculaires.

🫀 INFOGRAPHIE DE SYNTHÈSE / CHAPTGPT
Inflammation et athérosclérose : du LDL à l'infarctus
(d’après Ajoolabady et al., Cell Death & Disease, 2024)
🧬 1. LE DÉCLENCHEUR : LE LDL S'ACCUMULE
🔴 Excès de LDL sanguin
⬇️
🔸 Rétention dans la paroi artérielle
🔸 Oxydation → LDL oxydé (oxLDL)
⬇️
🧲 Attraction des monocytes circulants
⬇️
🦠 Transformation en macrophages
🍔 2. NAISSANCE DES CELLULES MOUSSEUSES
🦠 Macrophages + oxLDL
⬇️
🍔 Cellules spumeuses (foam cells)
Conséquences :
🔥 Sécrétion de cytokines inflammatoires
(IL-1β, TNF-α, IL-6)
📣 Recrutement de nouvelles cellules immunitaires
📈 Croissance de la plaque
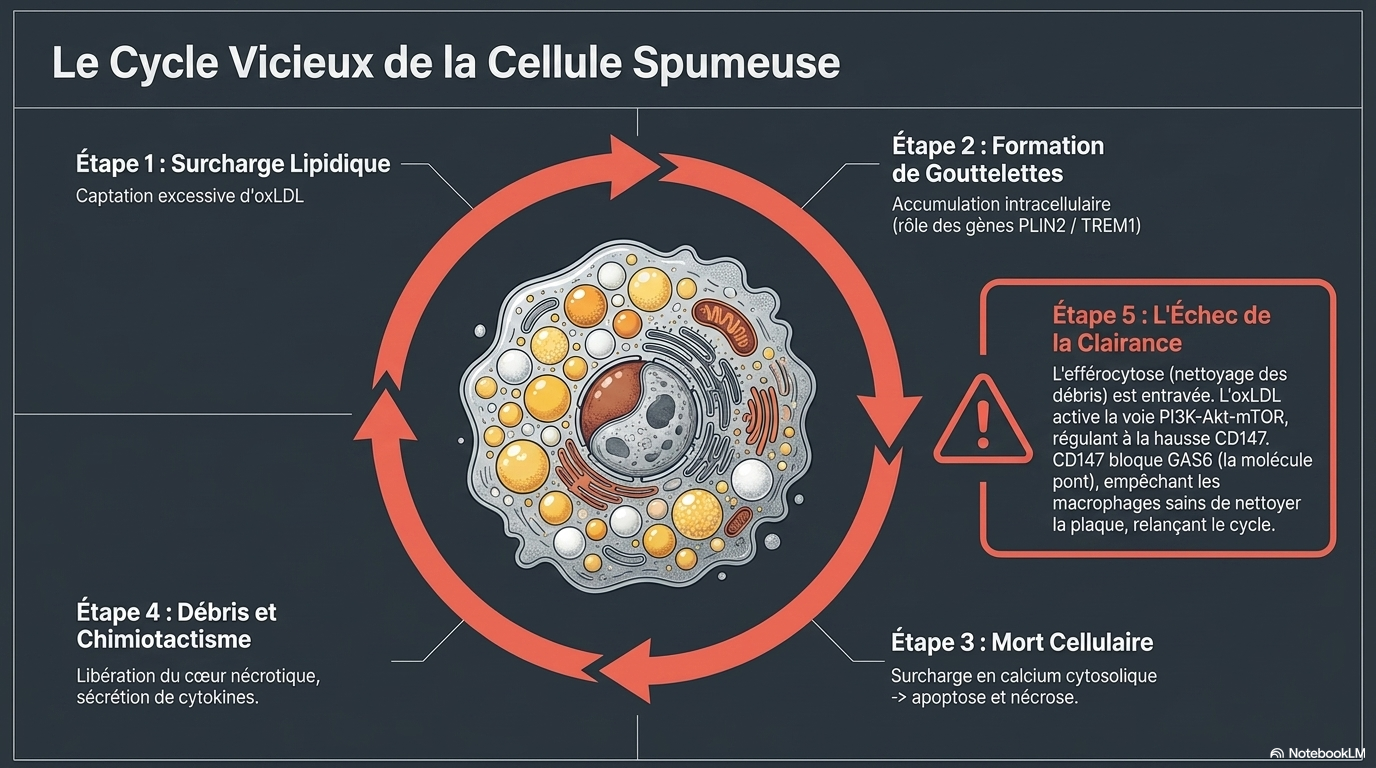
🔥 3. LE CERCLE VICIEUX INFLAMMATOIRE
oxLDL
↓
Macrophage
↓
Cellule mousseuse
↓
Inflammation
↓
Mort cellulaire
↓
Débris nécrotiques
↓
Nouvelle inflammation
↺Le cœur de la plaque devient un :
☠️ Noyau nécrotique inflammatoire
🧪 4. LES PRINCIPAUX MÉCANISMES MOLÉCULAIRES
🌬️ Hypoxie de la plaque
⬆️ HIF-1α
⬇️
🧬 VEGF
⬇️
🩸 Néovascularisation
⬇️
⚠️ Instabilité de plaque
💣 Inflammasome NLRP3
oxLDL / HMGB1
⬇️
NLRP3
⬇️
IL-1β + IL-18
⬇️
🔥 Amplification de l'inflammation
🎯 NF-κB : chef d'orchestre
Activation par :
✅ oxLDL
✅ Hypoxie
✅ Cytokines
✅ TSH et FSH
⬇️
Production massive de médiateurs inflammatoires
⚠️ 5. RUPTURE DE PLAQUE
Plaque inflammatoire
⬇️
🧱 Chape fibreuse amincie
⬇️
💥 Rupture
⬇️
🩸 Thrombose
⬇️
❤️ Infarctus du myocarde
ou
🧠 AVC
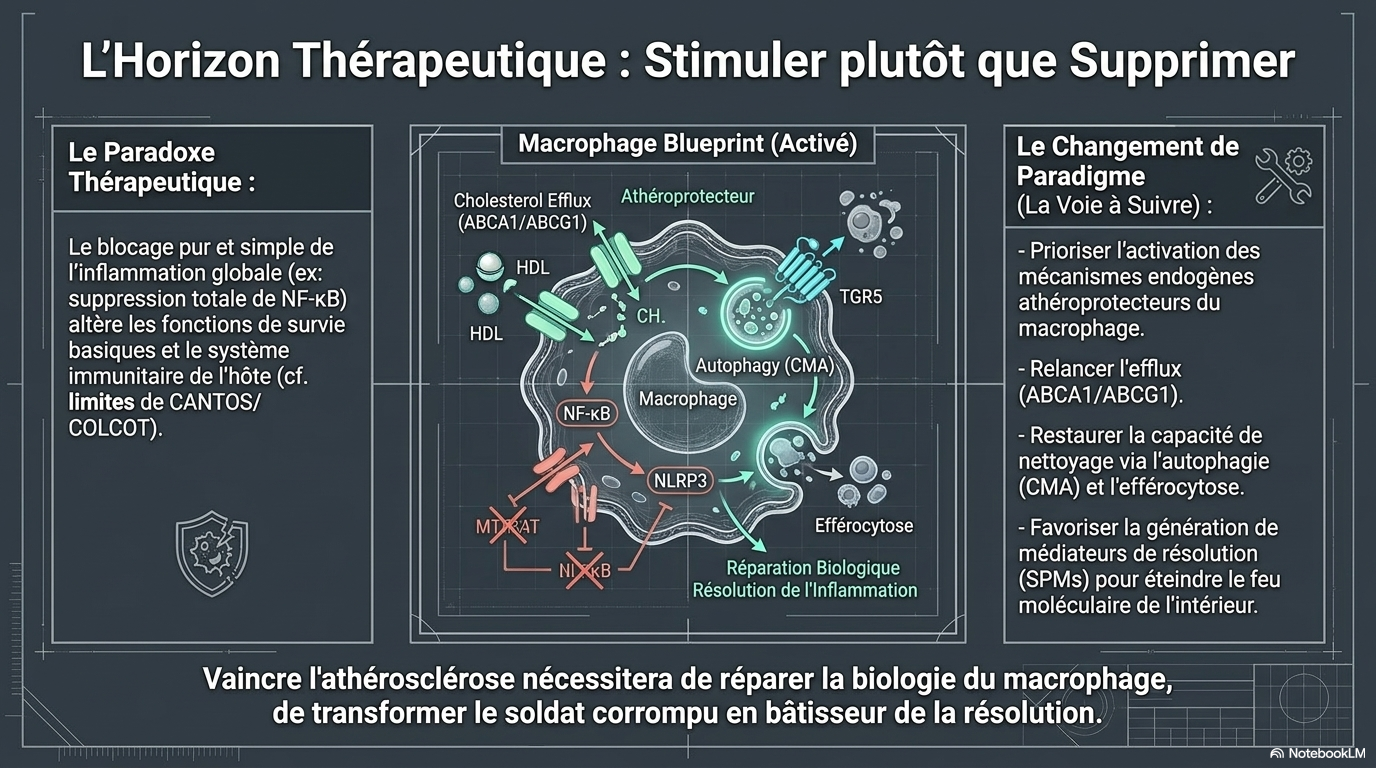
🛡️ 6. MÉCANISMES PROTECTEURS NATURELS
🔄 Efferocytose
Nettoyage des cellules mortes
➡️ Réduit l'inflammation
🚚 Transport inverse du cholestérol
ABCA1 / ABCG1
⬇️
HDL
⬇️
Foie
➡️ Élimination du cholestérol excédentaire
♻️ Autophagie
Nettoyage intracellulaire
➡️ Réduction des cellules mousseuses
🟢 Macrophages M2
Effets :
✓ Anti-inflammatoires
✓ Stabilisation des plaques
✓ Réparation tissulaire
💊 7. PISTES THÉRAPEUTIQUES
| Cible | Effet attendu |
|---|---|
| IL-1β (Canakinumab) | ↓ événements CV |
| Colchicine | ↓ inflammation résiduelle |
| TGR5 | Inhibition de NF-κB |
| NLRP3 | Cible prometteuse |
| Macrophages M2 | Stabilisation des plaques |
| Autophagie | Réduction des foam cells |
🎯 MESSAGE CLÉ
« L'athérosclérose n'est pas seulement une maladie du cholestérol ; c'est une maladie inflammatoire chronique de la paroi artérielle. »
🧬 LDL oxydé → macrophages → inflammation → rupture de plaque → infarctus/AVC.
La maîtrise de l'inflammation constitue aujourd'hui l'un des axes majeurs de prévention cardiovasculaire.
L'inflammation n'est peut-être pas un risque résiduel mais un risque immédiat dans la genèse de l'athérosclérose.
Alors pourquoi ne pas débuter immédiatement la colchicine simultanément qu'une statine et un antiagrégant plaquettaire ?
Autre vision possible , détecter l'athérome infraclinique le plus tôt possible, prévention primaire, et instaurer d'emblée la colchicine ou un autre anti-inflammatoire à visée vasculaire.
Il est peut-être temps de mettre en place un nouveau paradigme thérapeutique dans la lutte contre l'athérosclérose, ou alors la grande idée de l’inflammation est-elle une si bonne idée ?
Attention, ce n'est pas en regardant des témoins biologiques de l'inflammation vasculaire que l'on résout tous les problèmes !
La science est remplie de contradictions certes, mais elle avance toujours, les vérités scientifiques ont une durée de vie plus courte que la durée de vie des patients !...la vérité se cache dans les détails !
PERSPECTIVES FUTURES by OPEN EVIDENCE
Les essais en cours explorent de nouvelles cibles thérapeutiques incluant :
-
Inhibiteurs directs de l'inflammasome NLRP3 : Potentiellement bénéfiques pour l'ischémie coronarienne chronique et aiguë, ainsi que pour la stéatohépatite non alcoolique et la maladie rénale sévère [12]
-
Anakinra (antagoniste du récepteur IL-1) : Études en cours dans l'infarctus aigu [11]
-
Golodacimab (ciblant le récepteur LOX-1 des LDL oxydées) [11]
-
Rituximab (ciblant CD20 sur les lymphocytes B) [11]
Recommandations cliniques actuelles
Pour les patients avec athérosclérose établie et risque inflammatoire résiduel (CRP-hs ≥2 mg/L) :
-
La colchicine 0,5 mg/jour est l'option préférable compte tenu de son coût raisonnable et de ses preuves cohérentes [10]
-
Particulièrement indiquée chez les patients avec infarctus du myocarde récent ou maladie coronarienne stable chronique [1][3]
-
Risque modeste d'infections à surveiller
INFLAMMATION et ATHÉROSCLÉROSE : suite